
ComENet计算二面角的方式与GemNet、SphereNet等模型不同,它是从边的角度出发的。在GemNet和SphereNet等模型中,通常是从原子的立体结构和位置出发,使用球坐标系统来建模分子的几何结构。GemNet直接计算原子间的距离、夹角和二面角,依赖于对原子三维坐标的精确描述;而SphereNet则通过投影的方式对二面角的计算进行的简化。
ComENet则采用了一种更为抽象的方法,其专注于边的角度。ComENet首先定义了分子结构中的边,然后利用这些边来构建一个图的局部表示。在这种表示中,二面角不是直接通过计算四个原子的空间位置来得到的,而是通过分析连接这些原子的边之间的相对方向和转动。这种方法有助于模型更好地理解和预测分子在不同化学环境中的行为,因为它抓住了化学键之间相互作用的本质,具体方式如图1所示。
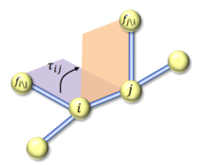
如图1所示,对于图中的任意一条边\(e_{ij}\),选取两个参考节点\(f_j^i\)和\(f_i^j\),分别代表除了节点j之外节点i的最近邻节点,以及除了节点i之外节点j的最近邻节点,\(\mathcal{I}_{ij}\)是从由\(f_i^j,i,j\)形成的平面到由\(f_j^i,j,i\)形成的平面的角度。
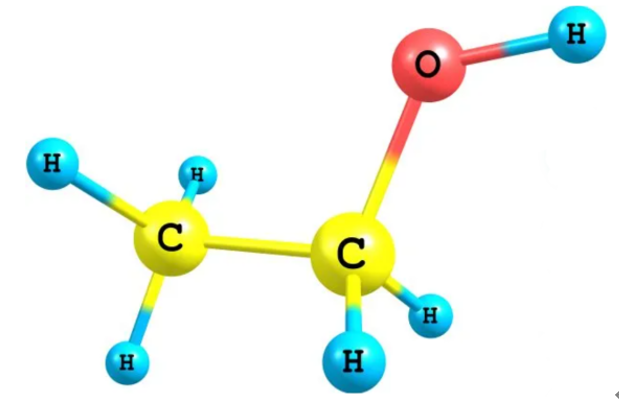
这种计算方式的侧重点是边的相对方向而非原子的绝对位置,因此降低了对计算资源的需求。具体来说,在传统的计算方法中,二面角的计算需要四个原子组合参与。而针对一个分子结构而言,四个原子的组合方法要远远多于边的数量,因此ComENet实际上是通过减少计算次数来提高计算效率的。以乙醇(Ethanol)分子为例,如图2所示。
其分子式为 C2H5OH,包含两个碳原子、一个氧原子,以及六个氢原子(共9个原子)。在乙醇中,主要的化学键包括碳-碳键、碳-氧键、以及碳和氧的氢键。具体连接如下:CH键5个、CC键1个、CO键1个、OH键1个,总共有8条边。但是,对于CH和OH边来说,H原子没有其他的邻居,ComENet二面角的计算方法并不适用,对于这种情况,通常会忽略这些边的转动角计算。实际上,单个氢原子的转动对分子的整体构象变化影响较小,尤其是在它只连接一个碳的情况下,因此这种情况下不计算边转动角也不会有太大影响。所以,实际参与二面角计算的边只有2个(CC键和CO键)。对比之下,如果是随机选择四个连续的原子进行二面角的计算,则共用12种,分别是H1-C1-C2-H4、H1-C1-C2-H5、H2-C1-C2-H4、H2-C1-C2-H5、H3-C1-C2-H4、H3-C1-C2-H5、H1-C1-C2-O、H2-C1-C2-O、H3-C1-C2-O、C1-C2-O-H6。对比之下,ComENet二面角的计算方法效率更高。
从另一个角度来说,ComENet计算二面角的过程中参与的四个原子是相互之间距离最近的,这使得模型能更有效地捕捉到分子内部的局部结构变化。这种局部信息特别重要,因为分子的化学性质和反应活性常常由其局部结构决定。因此,通过专注于这些局部连接的原子,ComENet不仅提高了计算效率,也增强了模型对分子行为的预测能力。
2 条评论
发表回复
作者
arwin.yu.98@gmail.com
相关文章
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
很好的博客,看完这篇博文后有两个问题
1. 乙醇分子例子中,计算CC键和CO键的二面角的时候,应该是10中而不是12种吧。
2. 没有理解为什么这里说ComENet二面角的计算方法效率更高,是说其他方法要穷举出上面所有的这些情况么,为什么不能随机选取一个进行计算呢,是计算出的所有二面角结果都会被使用么;如果是的话,ComENet的方法会导致一定的信息损失吧,我理解ComENet是在其中选了最重要的(距离最近的),在效率和损失之中做了trade-off?
计算CC键和CO键的二面角的时候确实是10种,谢谢指正!
至于问题2,我赞同你的观点,确实是在信息损失与计算量之间做的一种权衡,只不过这个权衡比较合理高效。