
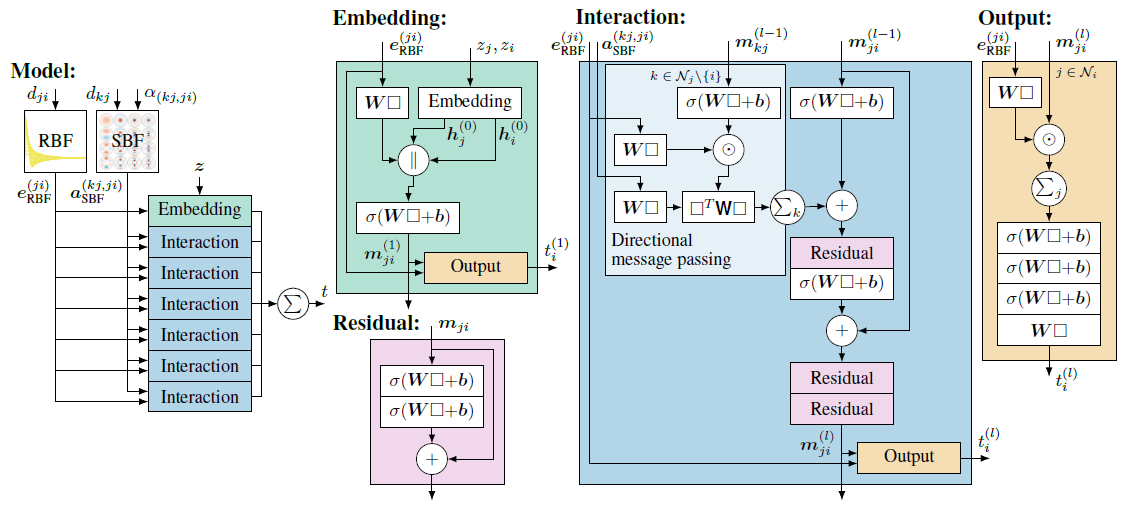
DimeNet(Directional Message Passing Neural Network)是一种专门用于分子图结构的神经网络,在预测分子性质和模拟分子间相互作用方面表现出色。DimeNet 的独特之处在于它引入了方向性消息传递机制,使其能够更好地捕捉分子中的几何和角度信息。DimeNet的关键特点之一是方向性消息传递,传统的消息传递神经网络(Message Passing Neural Network, MPNN)主要考虑节点和边之间的消息传递。SchNet进一步的对节点之前的相对距离进行建模。而DimeNet则更进一步引入了方向性消息传递的概念,这意味着它不仅考虑节点和边的信息,还将相邻边之间的角度信息纳入计算。
1.1引入角度信息
在SchNet模型中介绍的连续卷积中,存在一个截断距离C的概念。通过截断距离的设置,可以限制仅在这个距离范围内的原子之间才允许信息的传递。这种方法相当于在分子图中引入了一个局部邻域的概念,每个原子只与它的近邻原子“交流”,而不是与整个分子中的所有原子交流,这其实就是卷积算法中“关注局部”的思想与优点。
这种局部化的方法可以大大降低计算负担,因为它减少了模型必须处理的消息数量。同时,它也减少了模型可能学习到的噪声或不相关信息,因为远离的原子对通常对当前原子的化学性质影响较小。但是,这种方法也有其局限性,特别是当两个结构上不同但在局部尺度上相似的分子,例如在大约2埃的截断距离下,一个常规的GNN可能无法区分六边形分子(例如环己烷)和两个三角形分子(例如环丙烷)。这是因为在图神经网络(GNN)的常规处理方式中,原子之间的关系通常是基于它们之间的距离来建模的,而非它们在空间中的具体方向或者角度。当我们说一个原子的“邻域”时,是指在一定截断距离内与该原子相连的其他原子。在截断距离限制下,原子只能“看到”它周围一定范围内的其他原子,并与这些原子交换信息。
以环己烷(一个六边形分子)和环丙烷(一个由两个三角形组成的分子)为例,如果它们的键长相同,每个原子在截断距离内的邻域将是相似的,如图1所示。也就是说,每个原子周围可能都有两个相邻原子,它们之间的距离大约是2埃,因此从每个原子的局部视角来看,它们的邻域模式看起来是无法区分的。这样,基于局部信息的GNN将无法区分这两种分子的整体结构。
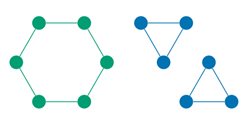
为了区分这两个分子,模型需要能够识别原子之间关系的角度特征,也就是需要知道这些原子是如何在三维空间中定向的。在三维空间中,六边形的角度和三角形的角度是不同的,因此这是区分这两种分子的关键。然而,如果模型仅仅基于距离信息而不考虑角度信息,它就会丧失这种区分能力。因此,为了更准确地建模分子结构,需要引入能够处理角度信息的机制,这便是DimeNet的设计动机。
1.2球谐函数和贝塞尔径向模型
在构建分子的基础表示时,DimeNet采用了球谐函数和贝塞尔径向模型这两类数学函数。球谐函数用于捕捉分子形状及原子在不同方向上的分布,这对于理解分子的三维结构至关重要。贝塞尔径向模型则在处理问题时更加关注于原子间的实际距离,这些函数特别适合于描述原子间距离相关的性质。
在DimeNet模型中,球谐函数和贝塞尔径向模型的应用是一种关键的数据预处理步骤,旨在从分子的三维结构中提取空间特征,尤其是原子间的距离和角度信息。具体来说,几何深度学习模型在处理原子问题时的输入一般是原子在笛卡尔坐标系(x,y,z)下的三维坐标。通过计算任意两个原子之间的欧几里得距离,并把这个距离作为贝塞尔径向模型的输入,贝塞尔径向模型能够捕捉到原子间距离上的波动特征。类似的,可以将笛卡尔坐标系下原子的位置表示转换为球坐标系中的角度表示,这些角度信息再作为球谐函数的输入,球谐函数能够捕捉到原子间方向上的角度特征。基于上述距离和角度,我们使用球谐函数和贝塞尔函数公式中的系数组合作为几何深度学习模型的输入特征。球谐函数和贝塞尔函数的数学特征正交性可以确保从这些转换得到的特征在信息上是独立且互补的,减少了特征间的冗余。
值得注意的是,球谐函数和贝塞尔方程公式中的系数是固定的数学函数,不需要被训练。只要有点的三维坐标作为输入,球谐函数和贝塞尔方程拆解的结果就是固定的。球谐函数和贝塞尔方程的使用更像是一种基于原子坐标的特征工程手段,用于将原始的几何数据转换成一种对模型更有用的形式。这种特征转换方法为DimeNet提供了一种“强大的语言”,使其能够精确描述并利用原子间的距离和角度相互作用信息。正交性保证了转换得到的特征集中每个特征都携带有用且独特的信息,有助于模型从数据中学习到更多独立的特征,从而提高预测分子性质的准确性。
最后,还有一个关键问题是模型如何习得这些系数特征的含义?毕竟球谐函数和贝塞尔函数并没有被内置在模型的结构中作为模型的一部分。实际上,DimeNet并不需要“知道”这些系数代表了什么。模型有能力通过训练过程学习这些转换后的系数特征与目标属性(如分子的能量或其他化学性质)之间的关系。通过大量的训练数据,DimeNet能够识别哪些模式(在这些系数表示的特征中)与预测的化学性质有关联。这种学习过程是数据驱动的,不需要模型对系数的物理意义有先验的理解。使用球谐函数和贝塞尔函数作为输入预处理步骤是因为它们提供了一种有效的方式来表达分子结构中的三维空间信息。这些信息是传统图神经网络(例如GCN、GraphSAGE、GAT等)难以直接处理的,因为它们通常设计用于处理图结构数据,而没有考虑到原子间复杂的空间和方向性关系。
DimeNet中使用的贝塞尔函数详见公式1:
\(\tilde{e}_{\mathrm{RBF},n}(d)=\sqrt{\frac2c}\frac{\sin(\frac{n\pi}cd)}d\)
公式1展示的是径向贝赛尔基函数\(\tilde{e}_{\mathrm{RBF},n}(d)\),这是一种只与原子间距离d有关的函数。其中n是索引,表示不同的基函数;c是截断距离,表示只有在此范围内的原子间相互作用才被考虑。这个函数是通过将正弦函数调整和缩放到[0,c]区间内得到的。这个区间是因为在分子模型中,通常只关心原子间在一定距离内的相互作用。正弦函数自然地产生了一个在两端为零且在中间振荡的形状,这个特性对于描述原子间距离上的波动特征是有用的。公式中的\(\sqrt{\frac2c}\frac{\sin(\frac{n\pi}cd)}d\) 是具体的函数形式,其中\(\sqrt{\frac2c}\)是为了确保函数在区间[0,c]上是归一化的,也就是说这个函数的值不会随着区间的大小而变换。

径向贝赛尔基函数曲线如图2所示,其中不同颜色的线代表不同的径向贝塞尔基函数。每条线对应于公式1中不同的n值,这些值代表了贝塞尔函数的不同阶。 n=1的基函数在整个区间内只有一个明显的峰值(d=0 时),并且在d=c 处下降至最小值0。这个性质与物理直觉一致:原子间相互作用在接近时最强,在远离时减弱,并在一定距离后消失。随着 d/c的增大,阶数n大于1的基函数会有更多的波峰和波谷。这可以表示在原子间距离上的变化更加复杂的模式。在某些情况下,随着距离的增加,原子间的相互作用可能会经历一系列的增强和减弱,这可能与分子内部原子的空间排列有关。正是这种复杂性使得基于贝塞尔函数的基函数成为捕捉和描述原子间相互作用特征的有力工具。
贝塞尔函数和球谐函数的结合方式的数学公式详见公式2,称为角向基函数:
\(\tilde{a}_{\mathrm{SBF},ln}(d,\alpha)=\sqrt{\frac2{c^3j_{l+1}^2(z_{ln})}}j_l(\frac{z_{ln}}cd)Y_l^0(\alpha)\)
公式2表示了如何结合贝赛尔函数\(j_{l}\)和球谐函数\(Y_{l}^0\)来构造一个球面傅里叶-贝赛尔基\(\tilde{a}_{\mathrm{sBF},ln}\),这个基函数用于分子内部原子间关系的建模。其中, \(j_1(\frac{z_{ln}}cd)\)是贝赛尔函数的l阶表示。\(Y_l^0\left(\alpha\right)\)是l阶的球谐函数,这里考虑的是m=0的特殊情况,只依赖于角度α。具体来说,球谐函数的一般形式为:
\(Y_l^m(\theta,\phi)=\sqrt{\frac{(2l+1)}{4\pi}\frac{(l-m)!}{(l+m)!}}P_l^m(\cos\theta)e^{im\phi}\)
其中:
(1)l是阶数(或角量子数),表示球谐函数的次数。
(2)m是磁量子数,范围是\(-l\leq m\leq l\)。
(3)\(P_l^m\)是缔合勒让德多项式。
(4)\(\theta \)是极角(或纬度),范围是\([0,\pi]\)。
(5)\(\phi \)是方位角(或经度),范围是\([0,2\pi]\)。
特殊情况:当m=0时,在这种情况下,球谐函数只依赖于角度\(\theta \):
\(Y_l^0(\theta)=\sqrt{\frac{(2l+1)}{4\pi}}P_l(\cos\theta)\)
其中\(P_l(\cos\theta)\)是勒让德多项式。
而公式2中的\(\sqrt{\frac{2}{c^{3}j_{l+1}^{2}(z_{ln})}}\)是归一化系数,用于确保不同l和n对应的基函数在给定范围内是正交的,并具有合适的规模。
为了帮助理解公式,对贝塞尔函数和球谐函数构成的球面傅里叶-贝塞尔基进行了可视化,如图3所示,其中每个圆圈代表了不同的l和n值下基函数的模式。
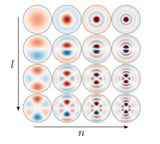
随着l值的增加,可以发现看到的是在角度上的变化越来越复杂;随着n值的增加,可以发现在径向上振荡的次数增加。图3中的这些圆形的子图是二维表示中的基础模式,可以用来表示原子间关系的多种可能性。通过组合这些基础模式,我们可以精确地描述原子之间的空间关系,并将这些信息输入到深度学习模型中进行进一步的处理和学习。这些基函数为分子的几何结构提供了一种数学描述,它既考虑到了原子间的距离,也考虑到了它们之间的角度,因此能够在不同的尺度和方向上分析分子。
总的来说,公式1和公式2提供了关于角度和距离的不同信息,并且被设计用于几何深度学习模型中的不同目的。公式2使用贝塞尔函数和球谐函数的组合来同时捕获距离和角度上的信息,这种表示在考虑原子间的相互作用时非常有用,因为这些相互作用通常取决于它们之间的距离和它们相对于彼此的方向。这个复合基础能够提供一个综合视图,表征原子如何在空间中相对于其他原子排列。公式1则是纯粹的径向基函数,它只关注原子之间的距离,而不考虑角度。这种表示提供了分子大小和形状的更宏观的视角,对于那些只依赖于距离的属性(例如某些类型的力)特别有用。
在后续的DimeNet模型结构中,网络层可以利用这些不同的信息来学习预测分子的性质,综合使用这些基函数可以让模型更全面地理解分子内部的复杂相互作用,提高预测的准确性和鲁棒性,从而在各自领域提供最佳性能。
1.3 DimeNet的消息传递机制
在 DimeNet 之前,几何图神经网络的网络结构最多只利用了几何信息中的距离信息,例如SchNet模型,而 DimeNet 是第一个将角度信息引入消息传递阶段的,其消息传递机制如图4所示。
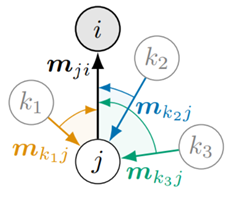
(1)节点:图4中的圆圈代表原子,标记为i和j,这些原子在分子图中通过边相连。
(2)消息:方向性嵌入\(m_{ji}\)被视为从原子j发送给原子i的一条消息。
(3)边:箭头表示消息的方向,从一个原子到另一个原子。例如,\(m_{ji}\)表示从j到i的消息。
(4)邻居:\(N_{j}\)表示原子j的邻居集合。在图中,邻居被标记为\(k_1,k_2\text{和}k_3\)。每个邻居都向原子j发送它们的消息\(m_{kj}\)。
(5)聚合:原子i通过聚合来自j的所有邻居k的消息来更新\(m_{ji}\)的消息。
在图4-17中,我们的目标是根据节点i的邻居信息来更新节点i的特征。通俗的解释,可以设想节点i是一个国王,他将向子民收税。首先,国王向大臣(最近的一圈邻居,例如节点j)收税。大臣们收到命令后,向老百姓收税(k),大臣们汇集之后,再向国王缴税。当然这是一个简化的模型。不过,可以很清晰的看到,DimeNet的消息传递不仅涉及邻居信息,还涉及邻居的邻居。这种涉及两跳(2-hop)的消息传递机制使得 DimeNet 能够获取更多的局部信息,另一方面,其算力消耗也大幅增加。
DimeNet 是第一个将角度信息引入消息传递阶段的,它不仅包括了两个节点(比如说原子i和j)之间的直线距离,还包括了一个第三个节点k,形成了一个角度。这个角度是通过计算节点 i和节点j 之间的向量以及节点i 和节点k 之间的向量的夹角得到的。通过这种方式,当节点更新其表示时,它已经集成了其周围环境的详细几何结构信息,公式表述如下:
\(m_{ji}^{(l+1)}=f_{\mathrm{update}}(m_{ji}^{(l)},\sum_{k\in\mathcal{N}_{j}\in\{i\}}f_{\mathrm{int}}(m_{kj}^{(l)},e_{\mathrm{RBF}}^{(ji)},a_{\mathrm{SBF}}^{(kj,ji)}))\)
其中:
(1)\(m_{ji}^{(l+1)}\)是更新后的消息,传递从原子j到 i的信息。
(2)\(f_{\mathrm{update}}\)是消息更新函数。
(3)\(m_{ji}^{(l)}\)是当前层l的消息嵌入。
(4)\(\text{E}\)表示对j 的所有邻居k(除了i)进行求和。
(5)\(f_{\mathrm{int}}\)是内部消息整合函数。
(6)\(m_{kj}^{(l)}\)是从原子k到j 的当前消息嵌入。
(7)\(e_{\mathrm{RBF}}^{(ji)}\)是基于j和i之间的距离的径向基函数。
(8)\(a_{\mathrm{SBF}}^{(kj,ji)}\)是角向基函数,它包含了由原子k,j 和i形成的角度信息。
其中,径向基函数和角向基函数在1.2节均有介绍。\(f_{\mathrm{update}}\)的实现很简单,通过一个神经网络层进行映射学习即可,\(f_{\mathrm{int}}\)的实现详见公式6、7、8。
\(f_{\mathrm{int}}(m,d_{ji},d_{kj},\alpha_{(kj,ji)})=\sum_n[\sigma(\boldsymbol{W}m+b)]_nf_{\mathrm{filter}1,n}(d_{ji})f_{\mathrm{filter}2,n}(d_{kj},\alpha_{(kj,ji)})\)
\(f_{\text{filter}1,n}(d){=}(W_{\mathrm{RBF}}e_{\mathrm{RBF}}(d))_n\)
\(f_{\text{filter}2,n}(d,\alpha){=}(W_{\mathrm{SBF}}a_{\mathrm{SBF}}(d,\alpha))^TW_n\)
(1)交互函数\(f_{\mathrm{int}}\)
公式6描述了DimeNet模型中用于建模原子间交互的函数\(f_{\mathrm{int}}\)。这个函数的作用是捕捉原子i和j,以及一个第三个原子k之间的交互。对于每对原子间的交互,该函数计算一个表示交互强度的值,这个值通过将距离和角度信息与学习得到的滤波器\(f_{\text{filter}1,n}\)和\(f_{\text{filter}2,n}\)结合来实现。m代表的是与原子i相关的消息或特征, \(d_{ji}\)和\(d_{kj}\)代表原子i与j和原子k与j之间的距离,\(\alpha_{(kj,ji)}\)是由这三个原子形成的夹角。
函数内部的求和表示模型将所有这些不同的交互加权累积起来,其中\(\sigma\ )是激活函数,W和b是训练过程中学习的参数,这允许模型在处理每个原子对时对其交互强度进行编码。
(2)第一个滤波器\(f_{\text{filter}1,n}\)
该滤波器专注于处理原子间的距离信息。\(W_{\mathrm{RBF}}\)是权重矩阵,\(e_{\mathrm{RBF}}(d)\)是径向基函数的表示,它能将原子间的距离转换成一组能反映距离变化的特征。公式7表示:通过将距离d经过\(e_{\mathrm{RBF}}\)映射和与\(W_{\mathrm{RBF}}\)相乘,我们得到了反映该距离信息的特征。
(3)第一个滤波器\(f_{\text{filter}2,n}\)
与公式7不同的是,公式8考虑的不仅仅是距离,还有角度信息。\(W_{\mathrm{SBF}}\)和\(W_{\mathrm{n}}\)也是可学习的权重矩阵。\(a_{\mathrm{SBF}}(d,\alpha)\)是二维球形Fourier-Bessel表示,它将距离d和角度\(\alpha)\)结合起来转换为一组特征。这样做的好处是可以捕捉原子间更复杂的空间关系,例如原子如何围绕中心原子排列。
公式8说明了如何结合这些特征和权重来表示原子间的交互模式,当原子的位置或它们之间的角度发生变化时,交互特征也会发生相应变化。这是DimeNet模型能够理解分子几何结构并准确预测其性质的关键。
通过上述两个滤波器,DimeNet不仅捕捉了分子内各原子间的距离信息,还捕捉了原子之间复杂的角度信息,这样的处理方式是模仿分子内部如何实际发生相互作用的。DimeNet通过这些精心设计的滤波器能够捕捉复杂的空间依赖性。
1.4 模型结构
DimeNet模型的概览如图5所示,它展示了如何处理原子间的距离和角度信息以生成分子的最终嵌入表示t,详细的步骤如下:
1.距离和角度计算:
(1)对于任意一对原子i和j,计算它们之间的欧氏距离\(d_{ij}\)。
(2)对于涉及到三个原子i 、j和k的角度,计算它们形成的角\(\alpha(k,j,i)\)。
2.径向基函数(RBF)和角向基函数(SBF):
(1)使用径向基函数\(e_{\mathrm{RBF}}^{(j,i)}\)转换距离\(\boldsymbol{d}_{\hat{y}}\),这些函数将距离标量转换为高维的特征向量,以便神经网络能够更好地处理。
(2)使用角向基函数\(a_{\mathrm{SBF}}^{(k,j,i)}\)转换角度\(\alpha(k,j,i)\),这些函数帮助网络捕捉原子三元组之间的空间关系。
3. Embedding 层:
初始原子嵌入z(通常是通过原子类型或其他原子属性获得的特征向量)与由RBF和SBF处理过的距离和角度信息结合,进入Embedding层。这个层将初始的原子特征与结构信息整合在一起,产生更丰富的特征表示。
4. Interaction 层:
Embedding层的输出然后通过一系列的Interaction层。这些层通常包含了更深层次的神经网络结构,用于学习原子之间复杂的相互作用。
5. 输出:
经过一系列的Interaction层后,模型产生最终的输出 t,它是一个综合了所有输入信息的向量,这个向量可以用于预测分子的性质或作为其他下游任务的输入。
下面,依次解释每个子模块的详细结构和运算过程:
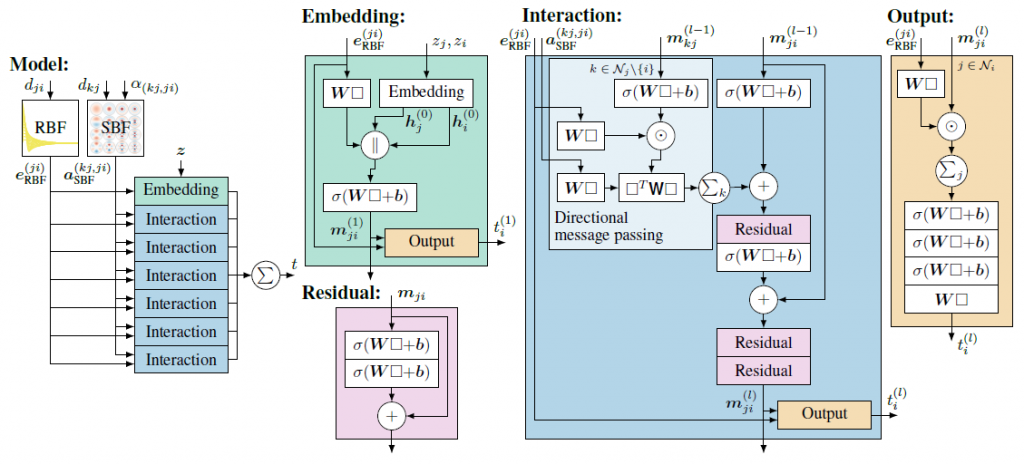
图神经网络中的嵌入(Embedding)部分如图6所示,嵌入步骤的作用是将原子的类型信息转化为丰富的特征表示,为进一步的消息传递和相互作用建模奠定基础,图6具体描述了原子间消息的嵌入生成流程。
\(z_{i}\)和\(z_{j}\)是两个原子的初始化特征向量,它们可以是根据原子的种类或其他化学性质随机初始化的,并通过后续的学习得以优化的表示。这些向量携带了原子的初始身份信息,是分子特性预测任务的起点。
对于原子j到原子i的每一对,它们的嵌入\(h_j^{(0)}\)和\(h_i^{(0)}\)被并列放置,与转换后的距离特征\(e_{\mathrm{RBF}}^{(j,i)}\)拼接在一起,形成了一个综合特征向量。然后,这个综合特征向量通过一个带权重矩阵W和偏置b的线性变换层,权重和偏置都是可通过训练得到的参数。通过这个变换,原子类型的离散特征与原子间距离的连续特征联合起来,映射到了一个新的特征空间。最后,激活函数\(\sigma \)被应用于线性层的输出,引入了非线性因素,这是神经网络能够捕捉复杂模式所必需的。这种非线性映射使得从一个原子到另一个原子的消息\(m_{ji}^{(1)}\)变得更加丰富,能够包含原子间更微妙的相互作用信息。
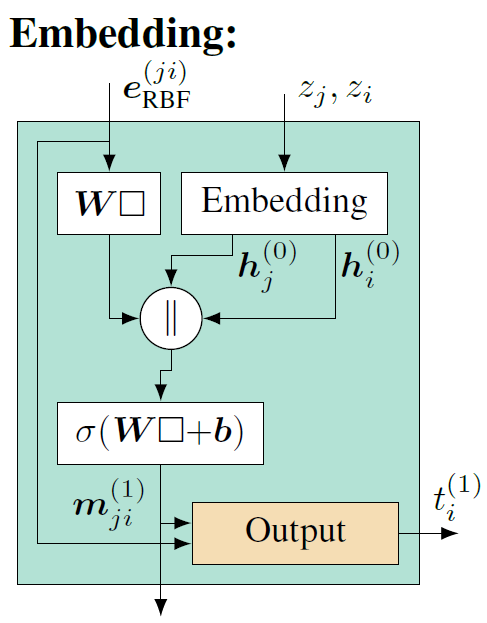
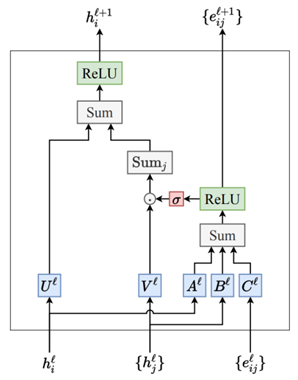
负责信息传递的交互层(Interaction)结构如图7所示。
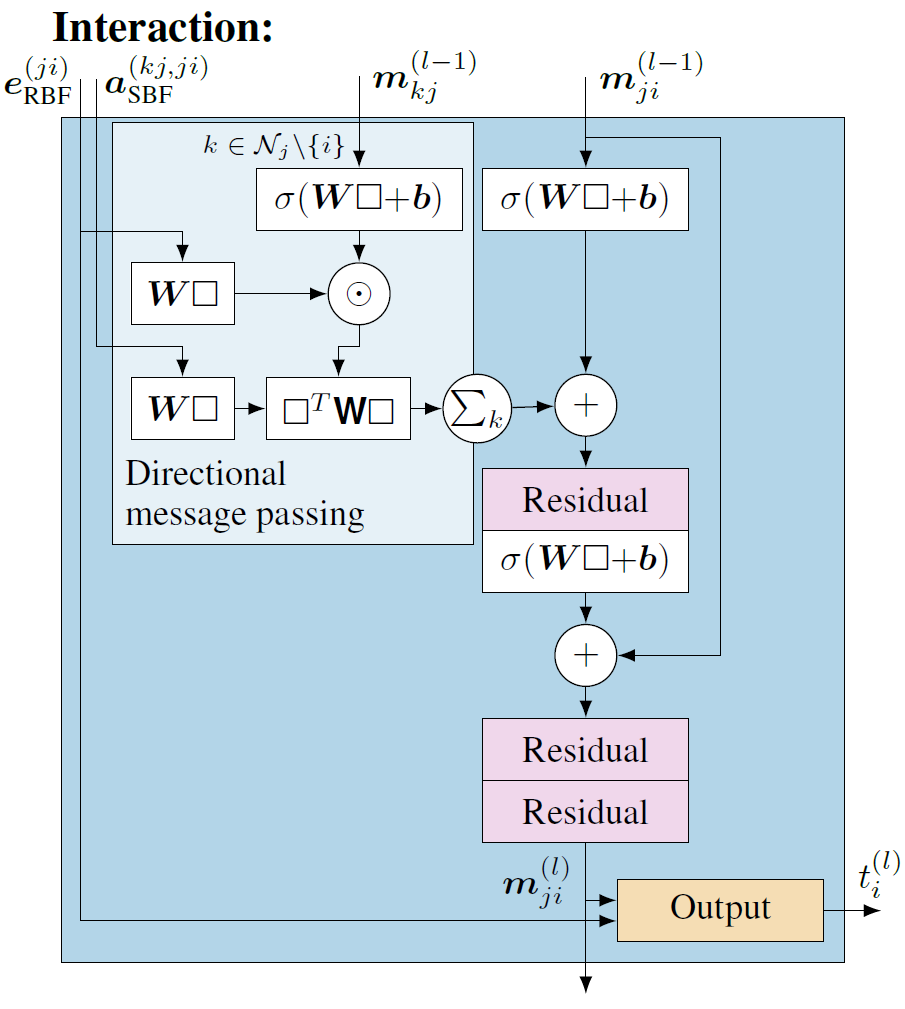
先解释图7中左上角的子部分,即方向性信息传递部分(Directional Message Passing)。输入\(e_{\mathrm{RBF}}^{(j,i)}\)代表使用径向基函数(RBF)编码的原子对之间的距离信息,捕捉原子之间的距离。而\(a_{\mathrm{SBF}}^{(k,j,i)}\)则编码了三个原子(i 、j和k)之间角度信息的球形基函数(SBF),它捕捉分子内部的三维空间关系。\(m_{kj}^{(l-1)}\)是从原子k到原子j的消息,是网络前一层(l-1 层)计算得到的。结合1.3节讲的消息传递, i是当前要更新信息的节点,j是i 的邻居,k是j的邻居( 邻居的邻居)。
径向基函数\( e_{\mathrm{RBF}}^{(j,i)}\)和球形基函数(a_{\mathrm{SBF}}^{(k,j,i)}\)的编码结果将分别经过一个线性变换,调整其表示,以便更好的与消息结合, 这里将两个线性变化后的结果称为tmp1和tmp2。\(m_{kj}^{(l-1)}\)包含更新节点i的邻居j和邻居的邻居k之间的角度信息的(详见1.3节),其结果将进一步通过一个带激活函数的神经网络层做进一步映射,结果称为tmp3。tmp3将与径向基函数\( e_{\mathrm{RBF}}^{(j,i)}\)线性变化后的结果tmp1(包含距离信息)进行元素间的乘法操作,得到的结果称为tmp4,tmp4能够捕捉两种类型的特征相结合后的复杂模式。
接下来\(\square^T\mathbf{W}\square \)是一个双线性层(Bilinear Layer),它通过对tmp2和tmp4进行独立的线性变换。tmp2表征的是基于球谐函数对i 、j和k之间角度信息的特征编码,tmp4表征的是融合了距离信息和经过非线性变换的邻居信息的结果。它们之间的交互不仅仅是简单的特征融合,而是一种能够表示特征对之间复杂关系的方法。这种高阶的特征交互在理解原子间如何相互作用时尤为重要,因为它们通常以复杂的方式相互依赖。最后∑k表示要综合考虑所有邻居的邻居信息,即两跳(2-hop)的消息传递机制,得到的中间结果称为tmp5。
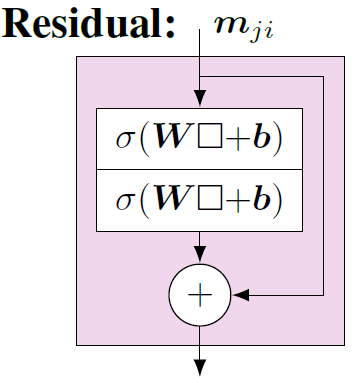
方向性信息传递部分(Directional Message Passing)以外的模型结构逻辑就相对比较简单清楚。首先,\(m_{ji}^{(l-1)}\)包含更新节点i 和其邻居j之间的角度信息,处理方式与\(m_{kj}^{(l-1)}\)一样,通过一个带激活函数的神经网络层做进一步映射。映射后得到的结果与tmp5相加,表征综合考虑更新节点i 的邻域信息和邻域的邻域信息,最后再经过一些神经网络层做进一步的学习映射,得到结果\(m_{ji}^{(l)}\)。在这些神经网络层的实现中,也加入了残差连接的实现,用于训练过程中更好的梯度信息传递,帮助模型收敛,残差模块结构如图8所示。
最后,交互层的结果\(m_{ji}^{(l)}\)不仅作为下一个交互模块的输入信息,也与\( e_{\mathrm{RBF}}^{(j,i)}\)一起送入该交互模块的顶层(Output层),并将顶层的计算结果作为当前交互模块的输出\(t_i^{(l)}\),顶层的模型结构设计如图9所示。
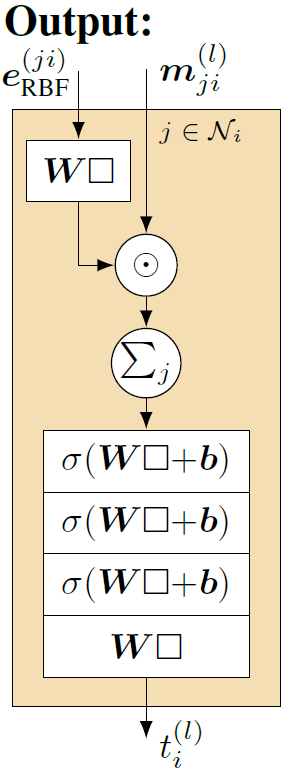
如图8所示,在交互模块的顶层设计中,包含节点对之间距离信息的\(e_{\mathrm{RBF}}^{(ji)}\)首先经过线性映射,然后与包含节点对角度信息的\(m_{ji}^{(l)}\)进行相乘求和,完成信息的交互与融合,最后再经过一些神经网络层的处理输入最终结果即可。
回顾总结整个模型设计,如图5所示,针对子模块Embedding和Interaction上述已经做了详细的介绍分析。DimeNet模型会取到每个交互模块的结果\(t_{i}\)进行求和得到最终的结果t。而且,距离信息RBF和角度信息SBF是贯穿整个DimeNet模型的,送入了每个交互模型,以帮助更好的进行节点间的信息传递,不仅仅是作为数据的预处理方式。这样做的好处是为了保持物理性质的一致性,即分子的物理属性是由原子间距离和角度确定的。如果距离和角度信息仅在第一层提供,那么随着信息在网络中的传递,这些物理属性的直接影响可能会逐渐减弱,从而影响模型的预测能力。在每个交互模块中重复这些信息确保了物理一致性。使其可以更全面地捕捉和学习分子内部复杂的三维空间结构和相互作用,从而提高了模型预测化学属性和反应的准确性。
1.5 DimeNet++
DimeNet++是DimeNet的改进版本,引入了一种更高效的消息传递机制。具体来说, DimeNet 通过利用方向性消息传递来考虑原子对之间的角度依赖性,这使得计算变得相对较慢和复杂。DimeNet++对此进行了改进,通过重新设计消息传递函数来减少计算量,同时保持了模型的表现力。此外,DimeNet++对模型的整体结构进行了简化,降低了模型复杂度,使得模型更易于训练,同时减少了过拟合的风险,以提高模型的泛化能力和预测性能。
相比于 DimeNet,DimeNet++在多个基准数据集上显示出了更好的性能,包括更高的准确率和更低的误差。简而言之,DimeNet++在保持DimeNet核心思想的基础上,通过对模型结构和计算过程的优化,实现了更高的效率和更好的性能,使其成为分子图表示和分子性质预测领域的一个强大工具。
DimeNet++的优化主要体现在交互模块(Interaction Block)中,回顾一下DimeNet的核心部件:方向性消息传递模块,如图10所示。
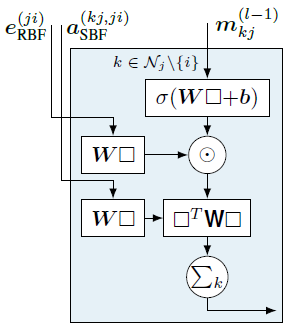
其中的关键是使用双线性层\(\square^T\mathbf{W}\square \)来进行嵌入\(m_{k,j}\)和基础表示\(e_{\mathrm{RBF}}^{(ji)}\)与\(a_{\mathrm{sBF}}^{(k,j,i)}\)间的信息交互,正如之前所说,这个层的计算非常昂贵,而且被用在模型最昂贵的方向性消息传递组件中。
DimeNet++用一个简单的哈达玛积(Hadamard Product,即元素间的乘积)和两个简单的神经网络层来替代原来的双线性层,以此来降低计算复杂度。为了补偿由于简化操作而可能带来的表达能力损失,增加了多层感知器(MLPs)来处理基础表示。例如,为径向基函数(RBF)和球形基函数(SBF)使用2层多层感知器(MLP)。这一改变在保持原始精度的同时显著降低了计算成本,改进后的方向性消息传递模块和交互模块如图11所示。
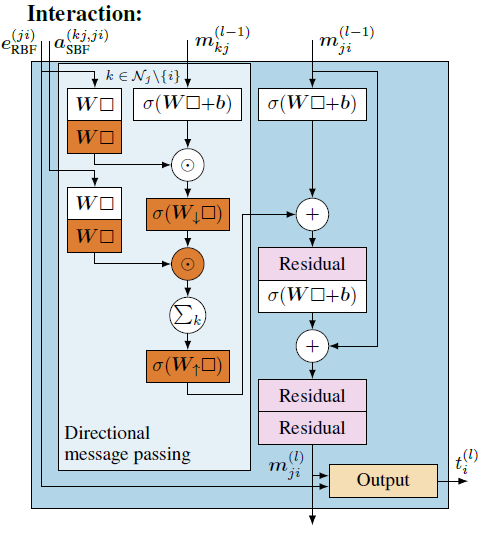
\(W_{\uparrow}\)和\(W_{\downarrow}\)是权重矩阵,用于在网络的不同层之间调整(上采样或下采样)特征的维度。 \(W_{\downarrow}\)通常用于减少特征维度,或者说是进行特征的下采样(downsampling)或维度的缩减。 \(W_{\uparrow}\)则相反,通常用于增加特征维度,即进行特征的上采样(upsampling)或维度的扩展。这样的层次结构可以有效地处理不同大小和不同分辨率的特征表示,这在处理具有多尺度结构或多种类型交互的复杂图时尤其重要。同时,也有助于在减少计算量和避免过度拟合之间取得平衡。
此外, DimeNet++也做了其他调参实验,例如,将交互模块从原来的6层减少到4层。将三元组嵌入的大小减少到64。将输出(原子)嵌入的大小增加到256等,消融实验的结果如表1所示。
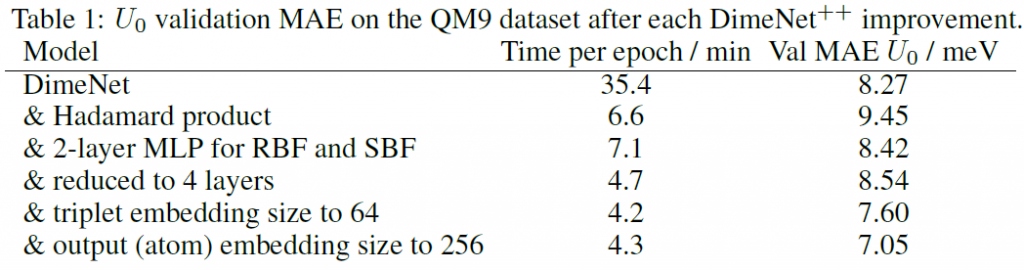
表1中列出了DimeNet及其在DimeNet++中各项改进后在QM9数据集上验证的平均绝对误差(MAE)以及每个epoch的训练时间。这些改进分别是:
(1)使用哈达玛积替换原有操作。
(2)为径向基函数(RBF)和球形基函数(SBF)使用2层多层感知器(MLP)。
(3)将模型层数减少到4层。
(4)将三元组嵌入的大小减少到64。
(5)将输出(原子)嵌入的大小增加到256。
在表格1中的第一列表示模型在数据集上训练一轮所消耗的时间,而第二列中的MAE是一种衡量模型预测分子的电量与真实结果之间差距的一种方式,其值越小代表模型的预测结果越好。从表格中的数据可以发现:
(1)原始的DimeNet模型的预测结果和真值之间的MAE为8.27毫电子伏(meV),每个epoch的训练时间为35.4分钟。引入哈达玛积之后,MAE略微增加到9.45 meV,但每个epoch的时间大幅减少至6.6分钟。
(2)添加2层MLP后,MAE降低到8.42 meV,每个epoch的时间为7.1分钟。
(3)模型简化为4层后,MAE略有上升至8.54 meV,但每epoch时间进一步减少至4.7分钟。
(4)将三元组嵌入大小减少至64后,MAE降低到7.60 meV,时间降至4.2分钟。
(5)最后,将输出嵌入大小增加至256,使MAE进一步降低到7.05 meV,每epoch时间为4.3分钟。
这些结果展示了通过逐步改进DimeNet++,不仅减少了每个epoch的训练时间,还在多数情况下改善了模型在预测分子性质方面的精确度。特别是当使用哈达玛积替代原先的双线性层时,模型的训练时间得到了大幅度的缩短。
作者
arwin.yu.98@gmail.com
相关文章
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}